

Abstract
Background: Recurrent somatic mutations in patients with myelodysplastic syndromes (MDS) implicate several epigenetic regulators in the molecular pathobiology of these disorders. We hypothesized that identification of MDS subtypes defined by DNA methylation patterns could improve prognostication and enhance our understanding of MDS disease biology. Methylation assays that investigate regions of the genome beyond promoters may better inform which regulatory regions possess biologic importance in the development and progression of MDS. Additionally, computational methods that agnostically approach large methylation datasets are needed when attempting to classify clinically and genetically heterogeneous disease populations such as MDS.
Methods: To measure DNA methylation, we used bisulfite padlock probes (BSPP), a targeted enrichment technique that captures approximately 500,000 unique CpGs in regions known to contain differentially methylated regulatory regions. BSPP was performed on bone marrow mononuclear DNA from 141 treatment-naïve patients with MDS that were genetically and clinically annotated in prior studies (Bejar et al. N Engl J Med 2011 and J Clin Oncol 2012). We used Onco-GPS, a computational method that identifies shared underlying cellular states among heterogeneous sample populations in an unsupervised manner, to identify DNA methylation subtypes of MDS (Kim et al. Cell Systems 2017). This method uses non-negative matrix factorization (NMF) followed by consensus clustering of the resulting NMF components. The optimal number of NMF components and consensus clusters were chosen to maximize the cophenetic correlation coefficient. A Fisher's Exact test was used to test for enrichment of genetic lesions in specific clusters. Univariate and multivariable models of overall survival (OS) were constructed using cox proportional hazards regression. Variables with univariate p-values <0.2 were evaluated for inclusion in the final multivariable model and the final model was constructed using a forward and backward stepwise procedure based on the Aikake Information Criterion.
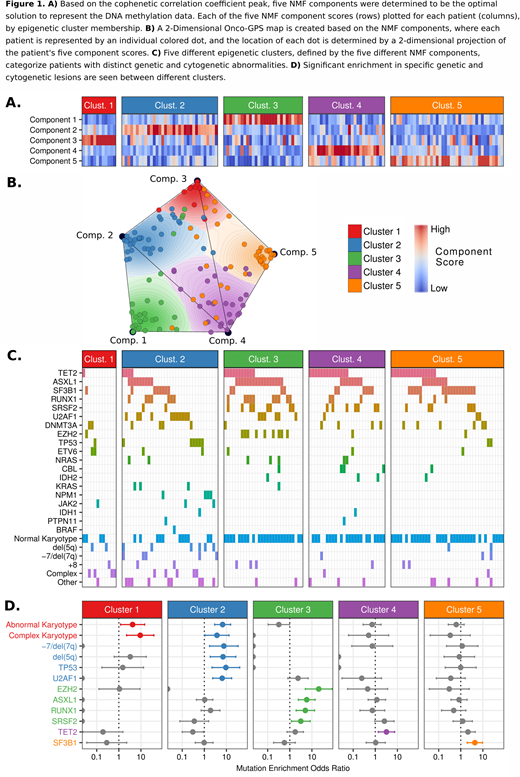
Results: We found that 5 NMF components and 5 consensus clusters maximized the cophenetic correlation coefficient, indicating the most stable partitioning of our dataset (Figures 1A-1B). The samples making up each cluster were enriched largely in a single NMF component, indicating that the underlying DNA methylation loci contributing to each cluster are distinct. Specific genetic and cytogenetic abnormalities were significantly enriched in different clusters, including enrichment of mutations in TP53 and U2AF1 in cluster 2; EZH2, ASXL1, RUNX1, SRSF2 in cluster 3; TET2 in cluster 4; SF3B1 in cluster 5; and abnormal and complex karyotypes enriched in clusters 1 and 2 (Figure 1C-1D). Kaplan-Meier curves revealed two major patterns of survival with clusters 2 and 3 displaying inferior OS when compared with clusters 1, 4, and 5. We combined clusters with similar median OS into "High Risk" and "Low Risk" cluster groups. In univariate Cox proportional hazards regression, High Risk and Low Risk cluster groups had significant differences in OS (HR, 1.95; 95% CI,1.24-3.08; p=0.003). In a multivariable model including elements of the IPSS-R, cluster risk group remained a significant predictor of OS (HR, 2.02; 95% CI, 1.25-3.27, p=0.004). We determined cluster-specific differentially methylated regions (DMRs) and found divergent patterns of methylation among clusters with the majority of DMRs located at non-promoter regulatory regions with high enrichment at both active and bivalent enhancers (as defined in a reference epigenome from mobilized CD34+ cells).
Conclusion: Using BSPP and novel computational methods, we found that DNA methylation of MDS patients identifies clinically relevant subtypes not otherwise distinguished by current prognostic models. Our analysis also shows that while genetic abnormalities can be associated with specific DNA methylation changes, patients with diverse genetic lesions can converge on common DNA methylation states, reflecting shared pathogenic mechanisms and clinical outcomes. Ongoing work to annotate the biological differences between the DNA methylation subgroups identified will enhance our understanding of the role of DNAm in prognosis and the underlying biology of MDS.
Zhang:Singlera Genomics: Consultancy, Equity Ownership, Membership on an entity's Board of Directors or advisory committees. Bejar:AbbVie/Genentech: Consultancy, Honoraria; Modus Outcomes: Consultancy; Takeda: Research Funding; Celgene: Consultancy, Honoraria; Astex/Otsuka: Consultancy, Honoraria; Genoptix: Consultancy; Foundation Medicine: Consultancy.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal